
Table of contents:
- BeStSel Tutorial
- BeStSel Updates and Bug fixes
- Practical considerations/guide for CD spectroscopy measurements
- BeStSel Q&A
BeStSel Tutorial
Click here to DOWNLOAD the BeStSel Tutorial (PDF, 2.1 MB)
BeStSel Updates and Bug fixes
Bug fixes, 18th March, 2024
We have fixed an issue with the Multiple spectra analysis modul. The modul worked only on data with 1 nm step. Now it can analyze CD spectra with 0.1, 0.2, 0.5 and 1 nm wavelength steps. Other issue fixed is that in the results, the abscissa was not correctly labeled.
Earlier, we have fixed an issue with the extinction coefficient calculator at 214 nm. It did not calculate correctly the extinction coefficient in those rare cases when the sequence started with proline residue, causing 1-2% error in the calculation for an average protein.
We thank our users for notifying us about these problems! Any feedbacks are welcome.
BeStSel webserver update, 13th September, 2023
The updated version of BeStSel (v1.3.230210) applies data smoothing with 2 nm window prior to secondary structure analysis. This smoothing window has no significant effect on the analyses of noiseless, smooth spectra. However, in the case of noisy spectra, which often occur in the lower wavelength range, this smoothing increases the performance of the method. In case our users do smoothing themselves, we do not recommend a stronger smoothing (i.e. using a larger window) because that will significantly distort the spectrum and affect the secondary structure estimation.
In general, we suggest to record the CD spectra with 0.1 nm data pitch in as high quality as possible by increasing the recording time of data points in step scan mode or increasing the number of accumulated scans in continuous scanning mode. Please, note that BeStSel will provide you the fitting with 1 nm data step.
NRMSD: In the case of noisy spectra, there will be always larger RMSD and NRMSD values, calculated between the input and the fitted spectra, than in the case of a noiseless spectrum, even if BeStSel has a similar performance. In such a situation, the RMSD and NRMSD values will be largely proportional to the noise and will not reflect correctly the reliability of the BeStSel fitting. To show how good the fitting is, instead of the input spectrum, we use the smoothed spectrum vs. the fitted one for NRMSD calculation. For RMSD, we kept the calculation between the original experimental spectrum and the fitted one, thus, RMSD is highly sensitive for noise level.
Multiple Spectra Analysis: now, the available wavelength ranges are the same as in Single Spectrum Analysis
We are grateful for any comments, bug reports, please, do not hesitate to write us at the Contact page or in e-mail.
Practical considerations/guide for CD spectroscopy measurements
You can find a more detailed protocol for CD measurements and data analysis in Micsonai et al. (Methods in Mol. Biol. 2021)
1. Sample preparation
a. Buffer composition
- Choose a buffer whose components have low absorption in the far-UV region. For a detailed list of commonly used buffer components and their absorption see (Micsonai, Bulyáki, and Kardos 2021). Phosphate buffer with the minimum amount of salt required for the protein is strongly preferred. For buffers with substantial absorbance in the far-UV use a cuvette with shorter pathlengths but be aware that this also requires higher protein concentrations.
- Avoid compounds with high absorption in the 180-260 nm range such as denaturing agents (GdnHCl and urea), reducing agents (dithiothreitol and mercaptoethanol) and organic solvents dimethyl sulfoxide (DMSO) or dimethylformamide (DMF). If necessary, use the denaturant dodine instead of GdnHcl or urea ( much lower concentrations of dodine are sufficient) and replace DTT and mercaptoethanol with Tris (2-carboxyethyl) phosphine (TCEP).
- Transfer the sample into the buffer to be used for the CD measurements either by dialysis or by using a centrifugal filter device. Dissolving lyophilized powder or diluting the protein from another buffer in the measurement buffer is not recommended as it can lead to contamination or a buffer composition differing from the reference buffer.
- Make sure that the protein sample is homogenous and free of contaminants, such as other proteins and chiral biomolecules (e.g. nucleotides).
- Check the purity of your sample by SDS-PAGE, reversed phase HPLC, mass spectrometry, absorption spectroscopy, or other complementary methods.
- Unless the aim is to study protein aggregates or amyloid fibrils, remove any precipitated or aggregated proteins by centrifuging at >10,000 x g. If needed, use ultracentrifugation at 100,000 x g to remove protein oligomers.
- When studying protein aggregation or amyloid fibrils, homogenize samples thoroughly by pipetting or using ultrasonication and apply only a short centrifugation at low force to remove large aggregates.
- Remove any tags or extensions from the protein of interest as these can affect the spectrum.
- If it is not possible to remove the tags, take their contribution into account when analyzing the data (number of residues, molecular weight, presumed contribution to the estimated secondary structure contents, possible effect on the secondary structure of the protein).
- It is essential to determine the protein concentration of the sample accurately!
- For proteins containing tryptophan and/or several tyrosine residues, concentration can be determined based on absorbance measured at 280 nm. The extinction coefficient can be obtained by using the ProtParam tool (https://web.expasy.org/protparam/).
- If the protein lacks tryptophan residues and contains no or very few tyrosine side chains, absorbance measured at 205 (Anthis and Clore 2013) or 214 nm (Kuipers and Gruppen 2007) should be used to calculate the protein concentration. The corresponding extinction coefficients can be retrieved from the BeStSel homepage (https://bestsel.elte.hu/extcoeff.php). The magnitude of the extinction coefficients at 205 and 214 nm makes it possible to determine the protein concentration directly on the CD sample.
- Quartz cells can be used for recording spectra above 180 nm. For low sample volumes or measurements that go below 180 nm use calcium fluoride cells.
- Select the pathlength of the cell so that the product of the pathlength in mm and the protein concentration in mg/ml is around 0.1.
- If the measurement buffer has substantial absorbance in the far UV region, choose a cuvette with shorter pathlength and increase the protein concentration according to the relationship provided above.
- To record spectra down to 180 nm on conventional instruments, use cells with short pathlengths (10-50 μm) and adjust the protein concentration accordingly.
- Make sure that the cell is clean and lint-free. Use a proper detergent and lint-free lens cleaning wipes to clean the cells before each measurement.
- The instrument should be calibrated regularly according to the manufacturer’s instructions.
- Let the UV lamp warm up for an hour after turning it on. Start measuring only after the lamp has warmed up.
- Collect data on the widest usable wavelength range from 260 nm down to at least 200 nm, but favorably to 190 or 180 nm. To measure CD data down to 175 nm use a synchrotron radiation CD (SRCD) instrument.
- Set the bandwidth to 1 nm or at most 2 nm.
- In continuous scanning mode choose a response or data integration time so that the wavelength is not shifted more than the value of the bandwidth during that time. (E.g. set response time to 0.5 sec when applying a scanning rate of 100 nm/min and 1 nm bandwidth.)
- Record and average several scans of each sample to achieve a better signal to noise ratio.
- Always record the baseline spectrum of the buffer using the very same settings as for the protein sample!
- Measure the CD spectrum of the buffer first. Estimate the usable wavelength range from the HT (high tension) values obtained, which should not exceed 50-60% of the maximum value provided by the manufacturer. Discard data measured at HT values above this limit.
a. Baseline subtraction
- Always correct the spectrum of the protein by subtracting the CD spectrum of the corresponding buffer (baseline)!
- Apply only moderate smoothing (Savitzky-Golay filtering) on the spectrum while taking care not to change any sharp component or steep part of the spectrum.
- The CD spectrum of the protein sample should be normalized for protein concentration, pathlength of the cell and the number of peptide bonds in the protein.
- If you use BeStSel for data analysis, you may upload the baseline-subtracted raw data and provide the required data on concentration, number of residues and pathlength and BeStSel will produce the normalized data for you (in Δε).
- Otherwise, normalize the baseline-corrected spectrum of the sample to obtain mean residue molar ellipticity [Θ]MRE in units of deg·cm2·dmol-1 according to the following relationship: [Θ]MRE = Θ/(10·c·Nr·l), where Θ is the measured ellipticity in mdeg, c stands for the molar concentration of the protein, Nr is the number of residues in the protein, and l is the pathlength in cm.
- If you prefer the use of Δε (M-1·cm-1), Δε=[Θ]MRE / 3298
References:
Anthis, Nicholas J., and G. Marius Clore. 2013. “Sequence-Specific Determination of Protein and Peptide Concentrations by Absorbance at 205 Nm.” Protein Science: A Publication of the Protein Society 22 (6): 851–58. https://doi.org/10.1002/pro.2253.
Kuipers, Bas J. H., and Harry Gruppen. 2007. “Prediction of Molar Extinction Coefficients of Proteins and Peptides Using UV Absorption of the Constituent Amino Acids at 214 Nm to Enable Quantitative Reverse Phase High-Performance Liquid Chromatography-Mass Spectrometry Analysis.” Journal of Agricultural and Food Chemistry 55 (14): 5445–51. https://doi.org/10.1021/jf070337l.
Micsonai, András, Éva Bulyáki, and József Kardos. 2021. “BeStSel: From Secondary Structure Analysis to Protein Fold Prediction by Circular Dichroism Spectroscopy.” Methods in Molecular Biology (Clifton, N.J.) 2199: 175–89. https://doi.org/10.1007/978-1-0716-0892-0_11.
BeStSel Q&A
What are the secondary structure basis components of BeStSel?
The secondary structure basis components of BeStSel are derived from DSSP. We introduced novel subgroups of the beta-sheets based on the beta-sheet twist. Parallel and antiparallel beta-sheets are distinguished and antiparallel beta-sheets are devided into three subgroups: left-hand twisted, relaxed, and right-hand twisted (anti1, anti2, anti3, respectively). The regular part of helices (helix1) and the distorted ends (helix2) are separated, similarly to SELCON3, however, only ?-helices are counted. BeStSel sorts 310-helix to "others". The definition of turn is identical to that in DSSP. The figure shows the eight basis components of BeStSel in relation to DSSP. For comparison, basis components of SELCON3 algorithm, which are also used for CONTIN and CDSSTR in CDPro, are presented.

How the fitting to the CD spectrum is carried out in BeStSel?
BeStSel fits the experimental CD curve by the linear combination of fixed basis components to get the proportion of the eight structural elements.
Considerations for wavelength range selection and the importance of the correct protein concentration?
BeStSel automatically offers the available wavelength ranges for calculations by inspecting the uploaded data. Choose the widest wavelength range for which the absorption (HT) is within the acceptable limit (cut-off).
The fitting reliability strongly depends on the correct protein concentration and cell pathlength, chosen to normalize the CD spectra. In case of uncertainty in the concentration and pathlength measures, a best correction factor may be calculated by BeStSel, providing the lowest rmsd. The calculation is efficient when using the wide wavelength range (175-250 nm). In case of correct normalization, fittings for different wavelength ranges should provide similar results.
How are the closest structures found in the entire PDB?
The BeStSel algorithm characterizes the secondary struncture of proteins by using eight components. Every single protein structure can be represented by a point in this eight dimensional secondary structural space. The distance between two points (x and x') is defined by their euclidean distance:
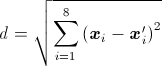
where xi is the content of the ith secondary structure of protein x. The search for the closest structures is carried out on the entire PDB of 71430 structures. The closest structures are presented by their PDB ID, secondary structure content, sequence length, number of chains and CATH domains contained. Links are provided for the PDB and CATH databases. This function is especially useful in case of multidomain proteins.
How are the closest single CATH domains found in the filtered PDB subset?
In case of single domain proteins, a prediction for CATH domains can be done. One option is the search for the closest structures on a single domain structure set. The definition of the distance is presented in the topic "How are the closest structures found in the entire PDB?". The single domain PDB subset is a non-redundant collection of chains containing single CATH domains or homodomains filtered for <=90% sequence homology and resolution better than 3.0 Å. This dataset contains 9218 domains covering 4 classes, 38 architectures, and 764 topologies.
The method of searching for the closest structures does not take into account the possible error of the secondary structure estimation. It can be used even if the secondary structural space is rarely populated by structures around the estimated point.
How are the structural domains found within the expected error of BeStSel?
The algorithm searches in a PDB subset with <= 90% sequential homology for all the chains having single CATH domains that lie within 1.5 × RMSD distance in each structural element from the estimated secondary structure. In other words we look up the structures in a box centered to the BeStSel result. The size of the box is determined by the RMSD of BeStSel on SP175 reference set. The hits in the box are sorted out for classes, architectures and topologies. The resulting table shows the frequencies and percentages of the different groups in the CATH categories in the order of frequency. In cases of architecture and topology the ten most populated groups are presented.
In the most dense regions of the secondary structural space hundreds of points can be found in the box. The advantage of the method is that the rarely populated folds located within the error of estimation that do not appear with the ""closest structure"" method will also be surveyed. In the case of some unique secondary structure composition no point or only a few points are located in the box. The closest structures method can be useful in such a situation.
On the analysis of peptide CD by BeStSel
BeStSel is optimized for proteins. However, it can work on peptides with the following considerations: Short peptides often have extreme amino acid composition very different from the average composition of proteins. In such cases, the possible side-chain spectral contributions can be distinct from that of proteins. Also, the structure of a peptide can be unique, which cannot be described by the basis spectra of BeStSel. As an example, the Turn component of BeStSel describes an "average turn", reflecting the various types of turns of proteins by a single basis spectrum. A very specific turn structure of a peptide can be different from the average turn basis spectrum of BeStSel. Similarly, the "Others" component, although mostly reflects irregular or unordered structures, is a collection of all the secondary structure elements excluded from the helix, beta and turn components.
As a kind of rule, when BeStSel fits the peptide CD spectrum well, we can assume the peptide spectrum is composed of components compatible with BeStSel and thus the fitting is reliable. If the fitting is imperfect (and this is not a concentration or normalization problem), the peptide CD spectrum probably has components shifted or different from that of BeStSel and the fitting might have larger errors. It is difficult to determine what NMRSD should be met for a reliable result. It is rather the spectral shape and catching of the spectral components that matters.